Molecular Dynamics Simulations
Molecular Dynamics Simulations deal with the simulation of materials (or mixtures of materials) on a molecular level. Even for a small simulation the number of particles can reach millions and, therefore, the computational effort is immense. Additional challenges arise if the molecules are not evenly distributed in the simulation domain. Therefore, efficient algorithms and parallelisation strategies are necessary to deal with this computational challenge. The focus of student work in this field is mainly on data structures, parallelisation and vectorization, but can also be in any other field related to molecular dynamics.
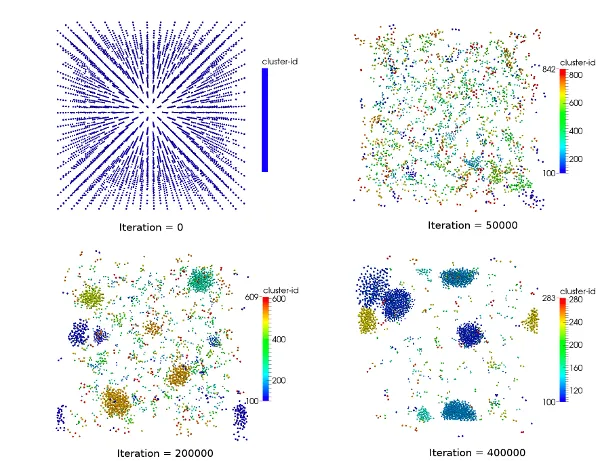
Examples for challenges in this field are:
- Load-balanced parallelization using space-filling curves and KD-trees
- Adaptive data structures for finding neighbour particles
- Improved parallelization schemes
- Efficient vectorization schemes
- Hybrid MPI+OpenMP parallelization
- Auto-Tuning
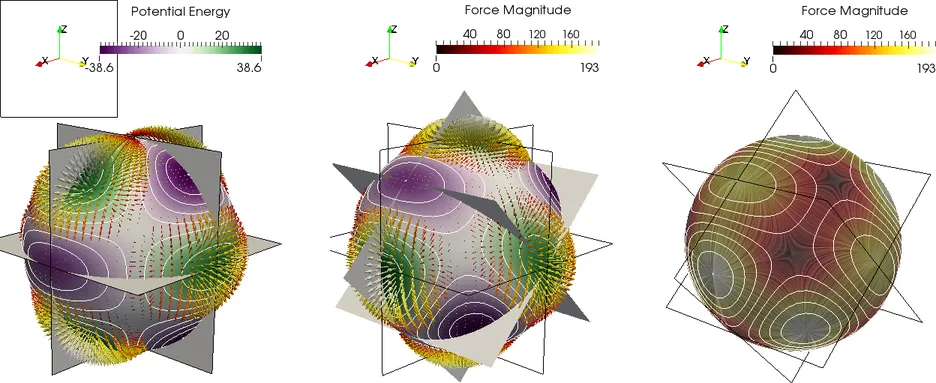
Further challenges arise when so called "long-range" interactions are present in the simulation. In such cases all interactions in the system need to be taken into account, leading to O(N^2) behavior. A different class of methods needs to be applied, such as the Barnes-Hut Method or the Fast Multipole Method, in order to solve the problem in O(Nlog N) or O(N) time.
Prerequisites:
- C/C++
- Linux
Contact:
- General Email for Particle Simulation Group: autopas(at)lists.lrz.de
- Samuel Newcome
- Markus Mühlhäußer
- Manish Mishra
- Jonas Schumacher